


My Talk at Harvard
Machine Learning Unveils the Language of Metabolites

This article is an adaptation of a recent talk I delivered at Harvard University in the Department of Chemistry and Chemical Biology. As the talk was not recorded, I felt it worthwhile to capture its essence in text.

Part 0: Preamble
Metabolites—the small molecules produced through metabolism—can be likened to individual words in a complex language that reflects an organism’s underlying metabolic state. Just as the presence, absence, and abundance of words in a sentence convey meaning, the intricate interplay of metabolites provides vital insights into health and disease. Advances in analytical chemistry tools, such as mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy, now enable the detection and precise measurement of these ‘words.’ However, interpreting this intricate metabolic language requires sophisticated analytical approaches. This is where machine learning (ML) plays a pivotal role, serving as a ‘translator’ by identifying patterns and relationships within metabolomic datasets to uncover insights into biological processes and disease states.
This essay will examine several applications of ML and AI in metabolomics research, highlighting examples from cancer biology, computational metabolomics, natural language processing (NLP), and how LLM-based agentic AI systems can help empower scientific (metabolomics) research.
Part I: Machine Learning Unveils Lipidome Remodeling Dynamics in Ovarian Cancer
[Bifarin, Sah et al. J. Proteome Res. 2023, 22, 6, 2092–2108]
[Previously recorded talk]
Ovarian cancer (OC) remains a significant challenge in women’s health, often diagnosed at advanced stages due to its asymptomatic early phases and vague later symptoms. High-grade serous ovarian cancer (HGSC), the most aggressive subtype, accounts for the majority of OC-related deaths. Understanding the metabolic dynamics of this disease, particularly in its early stages, is critical for developing effective diagnostic and prognostic tools.
To address this, we conducted a longitudinal study using the Dicer1-Pten double-knockout (DKO) mouse model, which closely mirrors the development and progression of human HGSC. Our research aimed to uncover temporal changes in the serum lipidome using machine learning (ML).
We collected biweekly blood samples from DKO and control mice starting at two months of age, analyzing them with ultra-high-performance liquid chromatography–mass spectrometry (UHPLC-MS). Given the differing lifespans of the two groups, direct comparison at equivalent chronological time points would be misleading. To resolve this, we employed a “percentage lifetime” metric, converting each mouse’s age at the time of blood collection into a percentage of its total lifespan. This approach enabled us to align the datasets and compare metabolic changes at equivalent stages of disease progression.
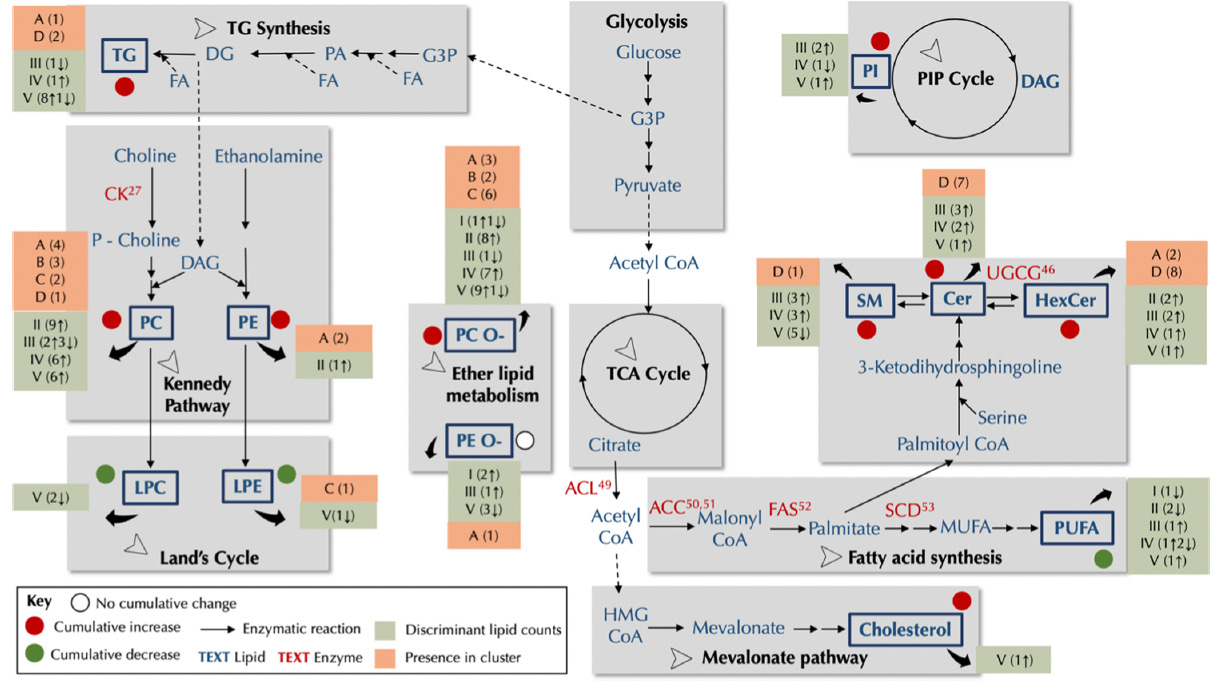
Global lipidome analysis revealed extensive remodeling.
Using a combination of supervised ML algorithms to distinguish between DKO and control mice at each life stage, and unsupervised ML techniques such as hierarchical clustering, we demonstrated that early HGSC progression is characterized by a marked increase in phosphatidylcholines (PCs) and ether PCs (PC-Os). This upregulation suggests a potential role for the Kennedy pathway in fortifying cell membranes during early cancer development.
As the disease progresses, we observe a shift toward more diverse lipid alterations. Notably, ceramides (Cers) and hexosylceramides (HexCers) show increased abundance in later stages. This finding aligns with the complex role of autophagy in cancer: initially acting as a tumor suppressor by clearing damaged cells, autophagy can later be co-opted by cancer cells to promote survival under metabolic stress. The observed increase in Cers and HexCers may reflect this functional shift in autophagy.
While the results provided valuable insights into the dynamics of the ovarian cancer lipidome and highlighted the potential of specific lipids as biomarkers, I figured the process of selecting and optimizing ML algorithms can be daunting for non-experts, and indeed be automated. This motivated my exploration of automated machine learning (Auto ML) and explainable AI (XAI) techniques to simplify the application of ML in metabolomics while simultaneously enhancing the interpretability of the resulting models.
Part II: Automated Machine Learning and Explainable AI for Metabolic Phenotyping
[Bifarin and Fernandez J. Am. Soc. Mass Spectrom. 2024, 35, 6, 1089–1100]
[Sah, Bifarin et al. Cancer Epidemiol Biomarkers Prev (2024) 33 (5): 681–693]
[Related Talk]
The application of ML in metabolomics faces challenges, especially for researchers without specialized ML expertise. Selecting the best-performing algorithms and tuning their hyperparameters requires significant knowledge and experience. AutoML frameworks, like Auto-sklearn, automate these crucial steps, streamlining the ML pipeline. However, the "black box" nature of many AutoML models can hinder interpretation and limit the extraction of biologically meaningful insights. To address this, I integrated XAI techniques, (Kernel SHAP) into the pipeline. I applied this AutoML-XAI approach to both kidney cancer urine metabolomics and OC serum lipidomics datasets.
Auto-sklearn consistently outperformed or matched standalone ML algorithms (Random Forest, SVM, k-NN) on key performance metrics, including AUC, accuracy, sensitivity, and specificity, in both datasets. This highlighted the effectiveness of Auto-sklearn's ensemble approach, which leverages the strengths of multiple algorithms and optimizes their hyperparameters for improved predictive performance.
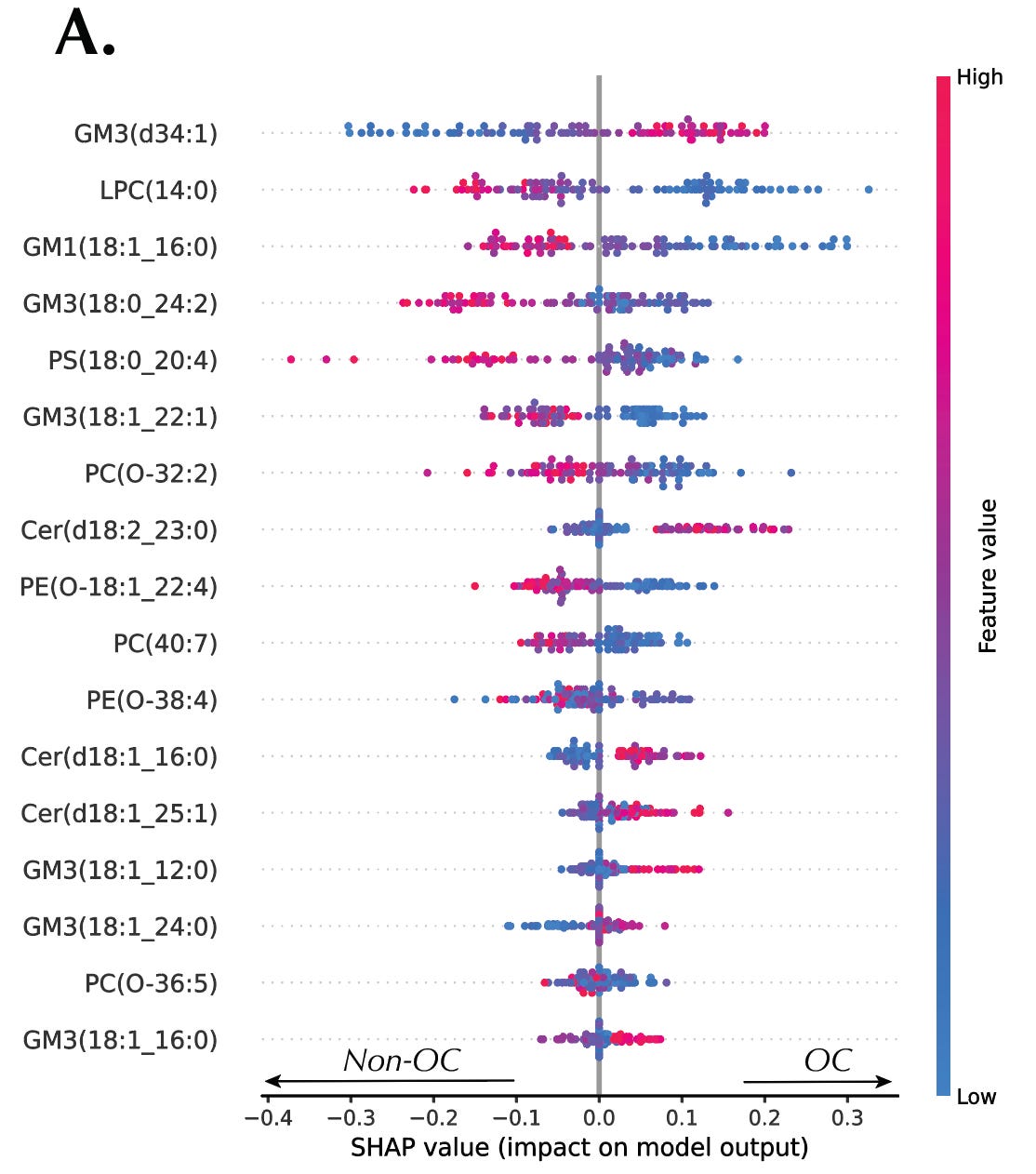
Kernel SHAP provided both global and local interpretations of the AutoML models. Globally, SHAP summary plots ranked the importance of each metabolite in driving the model’s predictions, identifying dibutylamine as the top discriminant feature in the RCC dataset and GM3(d34:1) in the OC dataset. Locally, SHAP waterfall plots visualized the contribution of each metabolite to individual predictions, offering detailed insight into how the model arrived at its decision for each sample. Furthermore, SHAP dependence plots revealed intricate interactions between metabolites. The interactions suggested potential links between metabolic pathways and processes.
Finally, SHAP decision plots facilitated error analysis by comparing feature importance between correctly and incorrectly classified samples. This allowed us to pinpoint which metabolites were most influential in driving misclassifications and refine our models accordingly.
Returning to the initial analogy, metabolites can be thought of as words, with ML acting as a translator to help us unravel the role of metabolites in various disease states (by mining the datasets). Similarly, we can view AI systems as linguistic analysts capable of exploring the entire corpus of metabolomics research, itself. In this context, the extensive body of published metabolomics literature represents an ongoing effort to decode the “language” of metabolism, with each paper contributing a new “chapter” to our collective understanding of altered metabolic processes. In the session that follows, I write about the application of Natural Language Processing (NLP) to map the landscape of metabolomics research.
Part III: Mapping the Landscape of Metabolomics Research
[Website: metascape.streamlit.app]
The exponential growth of scientific publications poses a significant challenge for researchers. Staying current with even a sub-field's literature has become incredibly difficult, and metabolomics is no exception. This information overload hinders our ability to synthesize existing knowledge, identify emerging trends, and pinpoint promising areas for future research. To address this challenge, we are applying state-of-the-art NLP-based techniques to map the evolving landscape of metabolomics research.
Our approach uses PubMedBERT embeddings of metabolomics abstracts, reduced in dimensionality with t-SNE and clustered using HDBSCAN. We leverage c-TF-IDF to identify characteristic keywords for each cluster, effectively summarizing the core topics within each research area.
Critically, we utilize LLMs to refine these keyword lists into concise, human-readable topic labels. By engineering specific prompts that include representative abstracts and keywords, we guide the LLM to generate descriptive labels in five words or less. This LLM-driven labeling significantly enhances the interpretability of the topic model compared to simply presenting lists of keywords.
The resulting interactive 2D embedding map reveals a nuanced landscape of metabolomics research, with distinct clusters representing diverse areas like plant stress responses, gut microbiota interactions, and cancer metabolism. We have developed a web app to democratize the research trend discovery, while we continue to write our paper and research ways to automate research trend discovery in-house.
Part IV: Empowering Metabolomics Research with Agentic AI
I concluded my talk by summarizing the core argument from my essay, “Artificializing Intelligence,” where I contended that the ongoing AI revolution signifies a profound shift in human progress, moving beyond the traditional view of AI as merely a predictive tool or chatbot. Instead, AI has begun to “artificialize” intelligence—at least some version of it—enabling transformative applications in science and knowledge work through agentic AI systems. These systems leverage iterative, reflective workflows, tool integration, and multi-agent collaboration to efficiently tackle complex tasks.
In the final segment of my talk, I revisited the key themes from the preceding three parts, generating pivotal questions from them and demonstrating how these questions can be addressed by designing LLM-based agentic AI systems.